 |
| Visit our sponsor at www.chemglass.com |
|
| Visit our sponsor at www.chemglass.com |
 |   |
| Hydroamination |
Contributed by:
Adam R. Johnson
Associate Professor of Chemistry
Harvey Mudd College
Claremont, CA, USA.
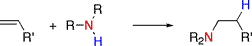
The hydroamination reaction is approximately thermodynamically neutral, but there is a high activation barrier due to the repulsion of the electron-rich substrate and the amine nucleophile. The hydroamination of alkynes is more thermodynamically favored than that of olefins, while allenes are intermediate in difficulty. The reaction has a high negative entropy (for the intermolecular reaction), making it unfavorable at high temperatures. As a result, catalysts are necessary for this reaction to proceed. Catalysts for the reaction include alkali metal bases, early and late transition metal complexes, gold, and lanthanide complexes. The field of hydroamination is evolving rapidly and has been reviewed regularly.1-7
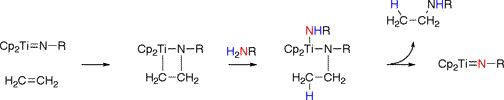
Lanthanide metal and cationic group IV metal catalysts react similarly. First a neutral or cationic amido complex forms which then undergoes a 1,2-insertion reaction on the unsaturated carbon-carbon bond to form a metal alkyl complex. The second part of the mechanism is similar to that described previously: addition of a second equivalent of amine substrate leads to protonolysis of the alkyl and regeneration of the amido complex.
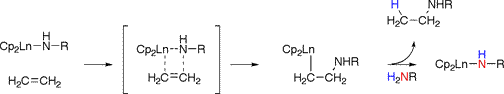
In contrast, late metal catalyzed hydroamination involves either activation of the amine group by the metal to form a hydrido-amido complex with subsequent reactions taking place at either the metal hydride or metal-nitrogen bond, or by activation of the unsaturated group by coordination to yield a more electrophilic group ready for nucleophilic substitution by an incoming amine.
Computational studies have helped to confirm and elaborate upon the basic mechanistic pictures described here.8-11
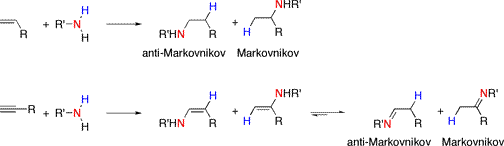
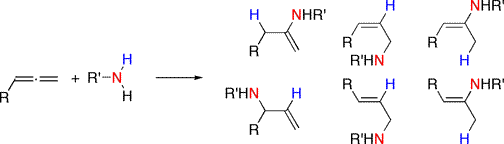
The intermolecular reaction with an unsymmetrical allene can yield many products; as a result, most hydroamination with allene substrates has been carried out in an intramolecular fashion. Often, the substrates for the olefin cyclizations require gem-dialkyl substitution which encourages preorganization via either a compression of the bond angle (the Thorpe-Ingold effect) or raising the energy of the ground state (the "reactive rotamer" effect.)
Intramolecular hydroamination results in cyclic products, as illustrated for the hydroamination-cyclization of an aminoolefin substrate with gem-dialkyl substituents.
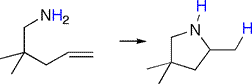
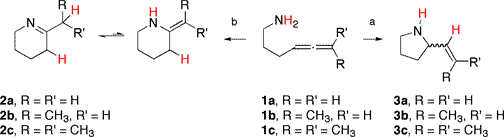
The intramolecular hydroamination of aminoallenes is potentially even more interesting, as the products are nitrogen containing heterocycles with a pendant vinyl group available for further reaction chemistry. Titanium amides, such as Ti(NMe2)4, are catalysts for the cyclization of the aminoallene to give the pyrrolidine products shown. The reaction is substrate dependent; when substrate 1a is used, only compound 2a is observed, while when 1c is used, only 3c is observed. Hydroamination of substrate 1b leads to a mixture of 2b and 3b (obtained as a mixture of cis and trans isomers). When chiral ligands are used, the product (3b and 3c,) can be obtained with an enantioselectivity of about 15%.15
Progress on the asymmetric cyclization of aminoallenes has focused on the use of gold complexes with bulky chiral phosphine complexes, resulting in high enantioselectivities (70-90 %ee).16-18
| Researcher | Location | Amination interests |
|---|---|---|
| Adam Johnson | Harvey Mudd College | Catalytic hydroamination chemistry of chiral titanium complexes |
| Aaron Odom | Michigan State | Titanium-based hydroamination catalysts |
| Laurel Schafer | University of British Columbia | Group 4 hydroamination catalysts |
| Ross Widenhoefer | Duke University | Pt-catalyzed olefin hydroamination and hydroalkoxylation |
| John Hartwig | University of Illinois at Urbana-Champaign | Pd-catalyzed olefin hydroamination |
| Kai Carsten Hultzsch | Rutgers University | Asymmetric hydroamination with lanthanides and lithium salts |
| Tobin Marks | Northwestern University | Actitinide and lanthanide catalysts |
This list is not exhaustive and any omissions of current researchers is simply an oversight. Contact us to add additional research groups.

[Index] [Keyword Search] [Books & Software] [ILPI Home Page]
Please visit our sponsor to thank them for supporting this site!
This page was last updated Tuesday, March 31, 2015
This document and associated figures are copyright 1996-2025 by Rob Toreki or the contributing author (if any) noted above. Send comments, kudos and suggestions to us by email. All rights reserved.